سرطان رتینوبلاستوماچیست و چگونه ایجاد می شود؟ – آروین کلینیک
سرطان زمانی شروع می شود که سلول ها شروع به رشد خارج از کنترل می کنند. سلولهای تقریباً در هر قسمت از بدن میتوانند به سرطان تبدیل شوند و به نواحی دیگر گسترش یابند. برای کسب اطلاعات بیشتر در مورد چگونگی شروع و گسترش سرطان، به مقاله سرطان چیست؟ برای اطلاعات در مورد تفاوت بین سرطان های دوران کودکی و سرطان بزرگسالان، به سرطان در کودکان مراجعه کنید .
رتینوبلاستوما سرطانی است که از شبکیه، قسمت پشتی چشم شروع می شود. این شایع ترین نوع سرطان چشم در کودکان است. به ندرت، کودکان ممکن است انواع دیگری از سرطان چشم، مانند مدولواپیتلیوما، که در زیر به اختصار توضیح داده شده است، یا ملانوم چشمی (چشم) داشته باشند .
برای درک سرطان رتینوبلاستوما ، دانستن نحوه عملکرد بخشهای چشم کمک می کند.
سرطان رتینوبلاستوما چگونه ایجاد می شود؟
چشم ها قبل از تولد شروع به رشد می کنند. در مراحل اولیه رشد، چشم ها دارای سلول هایی به نام رتینوبلاست هستند که با تکثیر سلول های جدیدی ایجاد می کنند که شبکیه را پر می کنند. در یک نقطه خاص، این سلول ها از تکثیر باز می ایستند و به سلول های بالغ شبکیه تبدیل می شوند.
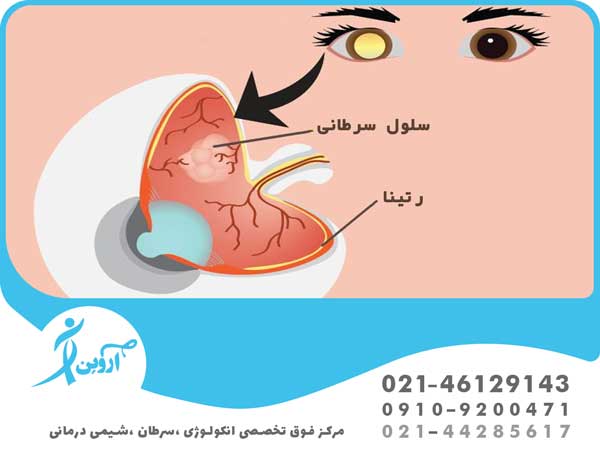
به ندرت اتفاقی در این روند رخ می دهد. به جای بلوغ، برخی از رتینوبلاست ها به رشد خارج از کنترل ادامه می دهند و سرطانی به نام رتینوبلاستوما را تشکیل می دهند.
زنجیره رویدادهای درون سلولی که منجر به سرطان رتینوبلاستوما می شود پیچیده است، اما تقریباً همیشه با تغییر (جهش) در ژن RB1 شروع می شود . ژن معمولی RB1 به جلوگیری از رشد خارج از کنترل سلول ها کمک می کند، اما تغییر در ژن مانع از عملکرد آن می شود. بسته به زمان و مکان تغییر در ژن RB1 ، می تواند منجر به 2 نوع مختلف رتینوبلاستوما شود.
تاثیر خصوصیات ژنتیکی بر سرطان رتینوبلاستوما
- رتینوبلاستوما اولین توموری بود که مستقیماً با یک ناهنجاری ژنتیکی (حذف یا جهش باند q14 کروموزوم 13) مرتبط بود.
- RB1 پروتئین pRb را رمزگذاری می کند که نقش کلیدی در چرخه سلولی بازی می کند: امکان تکثیر DNA و پیشرفت چرخه سلولی را فراهم می کند، زیرا در کنترل رونویسی ژن های فاز S شرکت می کند (G1 → †’S).
- علاوه بر سرطان رتینوبلاستوما ، ژن RB1 در سرطانهای مثانه ، سینه و ریه غیرفعال میشود .
سرطان رتینوبلاستوما ارثی
کودکان مبتلا به سرطان رتینوبلاستوما ارثی در سنین پایین تر از موارد پراکنده به این بیماری مبتلا می شوند. علاوه بر این، خطر ابتلا به سایر سرطانهای غیرچشمی در این کودکان افزایش مییابد، زیرا ناهنجاری در ژن RB1 مادرزادی است (یعنی از بدو تولد وجود دارد) و بر تمام سلولهای بدن (معروف به جهش ژرمین)، از جمله سلولهای هر دو چشم تأثیر میگذارد. سلول ها، شبکیه ها به همین دلیل، کودکان مبتلا به فرم ارثی اغلب به جای یک چشم، رتینوبلاستوما در هر دو طرف دارند.
علائم سرطان رتینوبلاستوما چیست؟
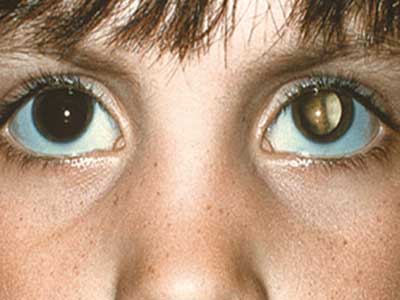
شایع ترین و واضح ترین علامت سرطان رتینوبلاستوما ، ظاهر غیرطبیعی مردمک است که در اثر برخورد پرتوی نور بازتابی به رنگ سفید متمایل به خاکستری دارد. سایر علائم و نشانه ها عبارتند از: کاهش بینایی، درد و قرمزی چشم و تاخیر در رشد. برخی از کودکان مبتلا به سرطان رتینوبلاستوما ممکن است دچار استرابیسم شوند (چشم هایی که در هم راستا نیستند). در موارد دیگر، امکان یافتن گلوکوم نئوواسکولار وجود دارد که پس از مدتی می تواند باعث بزرگ شدن چشم (بوفتالموس) شود.
سلول های سرطانی می توانند بیشتر به چشم و سایر ساختارها حمله کنند:
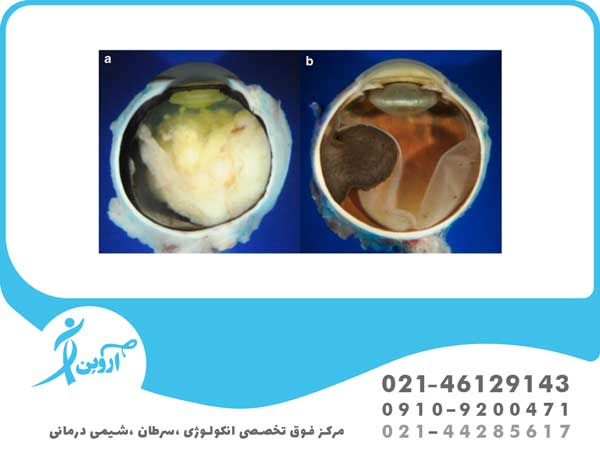
- رتینوبلاستوما داخل چشمی: زمانی که تومور به طور کامل در داخل چشم قرار گرفته باشد، سرطان رتینوبلاستوما را می توان به عنوان داخل چشمی تعریف کرد. نئوپلاسم فقط در شبکیه دیده می شود یا سایر قسمت ها مانند مشیمیه ، جسم مژگانی و بخشی از عصب بینایی را نیز تحت تأثیر قرار می دهد . بنابراین رتینوبلاستوما داخل چشمی به بافت های اطراف خارج چشم گسترش نیافته است.
- سرطان رتینوبلاستوما خارج چشمی: سرطان می تواند رشد کند تا بافت اطراف چشم را تحت تاثیر قرار دهد ( رتینوبلاستوم اربیتال ). سرطان همچنین می تواند به سایر نواحی بدن مانند مغز، ستون فقرات ، مغز استخوان و غدد لنفاوی ( رتینوبلاستوم متاستاتیک ) گسترش یابد.
تشخیص سرطان رتینوبلاستوما
- سونوگرافی مداری، CT یا MRI
- گاهی اوقات اسکن استخوان، آسپیراسیون مغز استخوان و بیوپسی، پونکسیون کمری
در صورت مشکوک بودن به تشخیص، هر دو فوندوس باید با افتالموسکوپی غیرمستقیم با مردمک های گشاد شده و کودک تحت بیهوشی عمومی به دقت بررسی شوند. تومورها در شبکیه به صورت برآمدگی های منفرد یا چندگانه خاکستری مایل به سفید ظاهر می شوند. منشا تومور ممکن است در زجاجیه قابل مشاهده باشد.
تشخیص سرطان رتینوبلاستوما معمولاً با سونوگرافی مداری، ام آر آی یا سی تی تایید می شود. تقریباً در تمام تومورها، CT می تواند کلسیفیکاسیون را نشان دهد. با این حال، اگر عصب بینایی در طول افتالموسکوپی غیر طبیعی به نظر برسد، MRI برای جستجوی گسترش تومور در عصب بینایی یا مشیمیه ترجیح داده می شود.
اگر مشکوک به گسترش عصب بینایی یا تهاجم گسترده مشیمیه باشد، باید سوراخ کمری و MRI مغز برای ارزیابی متاستازها انجام شود. از آنجایی که متاستاز از راه دور نادر است، ارزیابی مغز استخوان و اسکن استخوان ممکن است برای بیماران مبتلا به علائم استخوان در نظر گرفته شود.
کودکانی که والدین یا خواهر و برادری با سابقه رتینوبلاستوما دارند باید بلافاصله پس از تولد و پس از آن هر 4 ماه یکبار تا سن 4 سالگی توسط چشم پزشک ارزیابی شوند. بیماران مبتلا به رتینوبلاستوما نیاز به آزمایش ژنتیک مولکولی دارند و در صورت شناسایی جهش ژرمینال، والدین باید برای همان جهش آزمایش شوند. اگر نسل بعد از والدین حامل جهش در خط زایا باشد، باید همان آزمایشات ژنتیکی و معاینه چشمی منظم انجام شود. کاوشگرهای DNA نوترکیب می توانند به شناسایی حامل های بدون علامت کمک کنند.
درمان سرطان رتینوبلاستوما
هدف از درمان سرطان رتینوبلاستوما ، تلاش برای حفظ دید تا حد امکان مناسب است. تیم درمان باید شامل یک چشم پزشک اطفال با تجربه در زمینه رتینوبلاستوما، یک متخصص سرطان اطفال و یک متخصص رادیواکتیو باشد.
رتینوبلاستوما یک طرفه پیشرفته با انوکولاسیون کره با برداشتن تا حد امکان عصب بینایی برداشته می شود.
برای بیماران مبتلا به سرطان دو طرفه معمولاً می توان دید را حفظ کرد. گزینه ها شامل فتوکوآگولاسیون دو طرفه، شیمی درمانی داخل شریانی، یا انوکلاسیون یک طرفه و فتوکواگولاسیون، کرایوتراپی و پرتودرمانی چشم دیگر است. پرتو درمانی با تابش خارجی یا برای تومورهای بسیار کوچک با براکی تراپی (قرار دادن یک صفحه رادیواکتیو در دیواره چشم نزدیک تومور) انجام می شود.
شیمی درمانی سیستمیک، مانند کربوپلاتین، اتوپوزید، و وین کریستین، یا با سیکلوفسفامید به اضافه وین کریستین، ممکن است برای کاهش اندازه تومورهای بزرگ مفید باشد تا امکان استفاده از سایر درمان های اضافی (مانند کرایوتراپی، هیپرترمی لیزر) برای درمان سرطان فراهم شود. که دوطرفه هستند یا برای درمان سرطان هایی که به خارج از چشم گسترش یافته اند. با این حال، شیمی درمانی به تنهایی به ندرت می تواند این تومور را درمان کند.
معاینه چشمی هر دو چشم در فواصل 2-4 ماهه و در صورت لزوم یک دوره درمانی بیشتر توصیه می شود.